— FREE — Doping Functions¶
Header¶
- Files for the tutorial located in nextnano++\examples\basics
basics_1D_doping_predefined.in
basics_1D_doping_analytic.in (not compatible with the free version)
Introduction¶
This tutorial is the fifth in our introductory series. In the previous tutorials, we’ve already encountered one pre-defined doping profile - the constant one. In the following, we will see more possibilities to create doping profiles. After completing this tutorial, you will know more about:
different doping profiles, namely linear and Gaussian
crating custom doping profiles
Keywords: Gaussian1D{}, linear{}, import{ }
Overview¶
As an overview, Figure 2.4.17 shows all the structures that will be created in this tutorial.

Figure 2.4.17 shows doping profiles including linear and Gaussian functions (left) and user defined functions (right).¶
Using pre-defined doping profiles¶
In this example we demonstrate two pre-defined doping profiles, namely Gaussian and linear profiles. For that we consider the setup in Figure 2.4.17 (left). The associated input file is basics_1D_doping_predefined.in.
Specifying regions with dopants
37structure{ # this group is required in every input file
38 output_impurities{ boxes = yes} # output doping concentration [10^18 cm-3]
39
40 #---------
41 # material
42 #---------
43
44 region{
45 binary{ name = GaAs } # material: GaAs
46 contact{ name = whatever } # contact definition
47 everywhere{} # region spreads over the complete device
48 }
49
50 region{
51 binary{ name = InAs } # region: InAs
52 line{ x = [ 20.0, 30.0 ] } # position: x=20.0 nm to x=30.0 nm
53 }
54
55 #-------
56 # doping
57 #-------
58
59 region{
60 line{ x = [ 30.0, 40.0 ] } # position: x = 30.0 nm to 40.0 nm
61 doping{ # add doping to the region
62 gaussian1D{ # Gaussian doping concentration profile
63 name = "p-type" # name of impurity
64 conc = 1.0E18 # maximum of doping concentration [cm-3]
65 x = 35 # x coordinate of Gauss center
66 sigma_x = 1.0 # standard deviation in x direction
67 }
68 }
69 }
70
71 region{
72 line{ x = [ 0.0, 20.0 ] } # position: x = 0.0 nm to 20.0 nm
73 doping{ # add doping to the region
74 linear{ # linear doping concentration profile
75 name = "p-type" # impurity name
76 conc = [0, 6.0e17] # start and end value of doping concentration [cm-3]
77 x = [0.0, 20.0] # position: x=0.0 nm to x=20.0 nm
78 }
79 }
80 }
81}
We separated the structural set up in two sections: 1) material and 2) doping. In the doping section we use
linear{} and gaussian1D{} to specify the doping profiles. For defining the Gaussian profile
with the total doping concentration \(C_{conc}\), coordinate of the maximum \(x_0\) and standard deviation \(\sigma\), three parameters has to be specified. For defining the linear profile
we specify start and end value of doping concentration \([y_{start}, y_{end}]\) with the corresponding x coordinates \([x_{start}, x_{end}]\), both as vectors.
Specify impurity species
84impurities{ # required if doping exists
85 acceptor{ # select the species of dopants
86 name = "p-type" # select doping regions with name = "p-type"
87 energy = 0.045 # ionization energy of dopants
88 degeneracy = 4 # degeneracy of dopants
89 }
90}
Output
We simulate the device by clicking F8 on the keyboard. In the related output folder
you should find a plot of the concentration profile (\(\Rightarrow\) Structure \(\Rightarrow\) density_acceptor.dat) as shown in Figure 2.4.18.
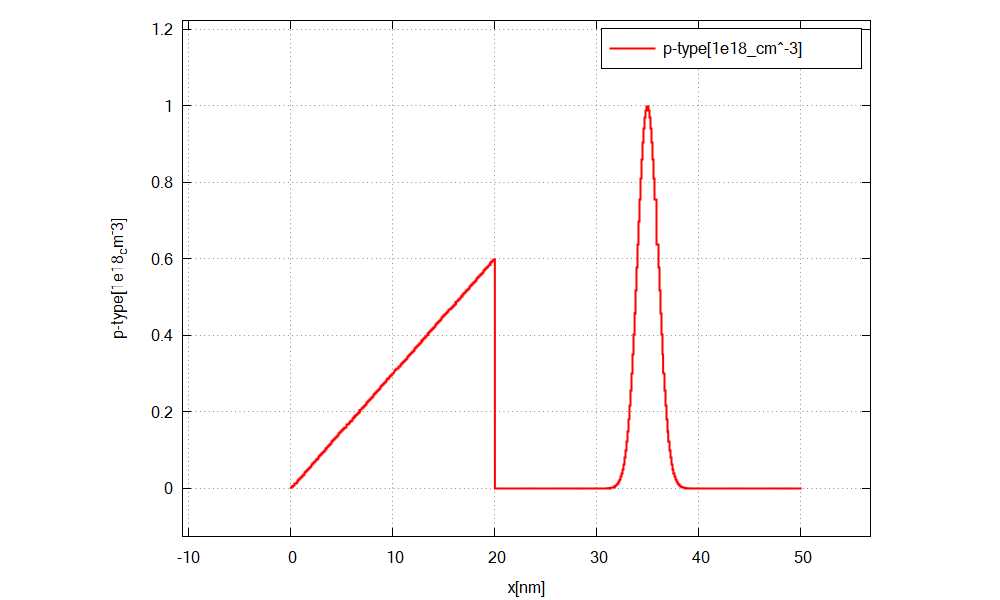
Figure 2.4.18 shows the doping concentration of donors along x.¶
2. Using custom doping profiles¶
In this example we introduce custom defined doping profiles. For that we consider the set up in Figure 2.4.17 (right). The associated input file is basics_1D_doping_analytic.in
Defining custom functions
20import{ # this group is optional
21 analytic_function{ # definition of analytic function
22 name = "custom_exp_fun_I" # name of function
23 function = "1e18 *(1-exp(-x+20))" # define the function
24 }
25 analytic_function{ # definition of analytic function
26 name = "custom_exp_fun_II" # name of fucntion
27 function = "1e18*exp(-x+30)" # define the function
28 }
29}
In order to create custom doping profiles, we have to define analytical functions in the group import{ } first. The analytical expression is
given by a string. Later, we can incorporate these functions for adding doping
by referring to the corresponding name.
Specifying regions with dopants
63structure{ # this group is required in every input file
64 output_impurities{ boxes = yes} # output doping concentration [10^18 cm-3]
65
66 #---------
67 # material
68 #---------
69
70 region{
71 binary{ name = GaAs } # material: GaAs
72 contact{ name = whatever } # contact definition
73 everywhere{} # region spreads over the complete device
74 }
75
76 region{
77 binary{ name = InAs } # region: InAs
78 line{ x = [ 20.0, 30.0 ] } # position: x=20.0 nm to x=30.0 nm
79 # overwrites the previously defined GaAs region
80 }
81
82 #-------
83 # doping
84 #-------
85
86 region{ # region: adds doping
87 line{ x = [ 20.0, 30.0 ] } # position: x=20.0 nm to x=30.0 nm
88 doping{
89 import{ # reference to import{ } group, where custom functions are defined
90 name = "n-type" # name of impurity
91 import_from = "custom_exp_fun_I" # import doping profile: custom_exp_fun_I
92 }
93 }
94 }
95
96 region{ # region: adds doping
97 line{ x = [ 30.0, 50.0 ] } # position: x=30.0 nm to x=50.0 nm
98 doping{
99 import{ # reference to import{ } group, where custom functions are defined
100 name = "n-type" # name of impurity
101 import_from = "custom_exp_fun_II" # import doping profile: custom_exp_fun_II
102 }
103 }
104 }
105}
Inside doping{}, the previously defined functions are used to create custom doping profiles. We
import each function (import_from) from the group import{ } by referring to the name that we had assigned.
The function is then evaluated on the interval specified inside line{} yielding the final doping profile.
Besides the shape of the doping profile we also specify the name, as usually.
Specify impurity species
108impurities{ # required if doping exists
109 acceptor{ # select the species of dopants
110 name = "p-type" # select doping regions with name = "p-type"
111 energy = 0.045 # ionization energy of dopants
112 degeneracy = 4 # degeneracy of dopants
113 }
114}
Output
We simulate the device by clicking F8 on the keyboard. In the related output folder
you should find a plot of the concentration profile (\(\Rightarrow\) Structure \(\Rightarrow\) density_donor.dat) as shown in Figure 2.4.19.
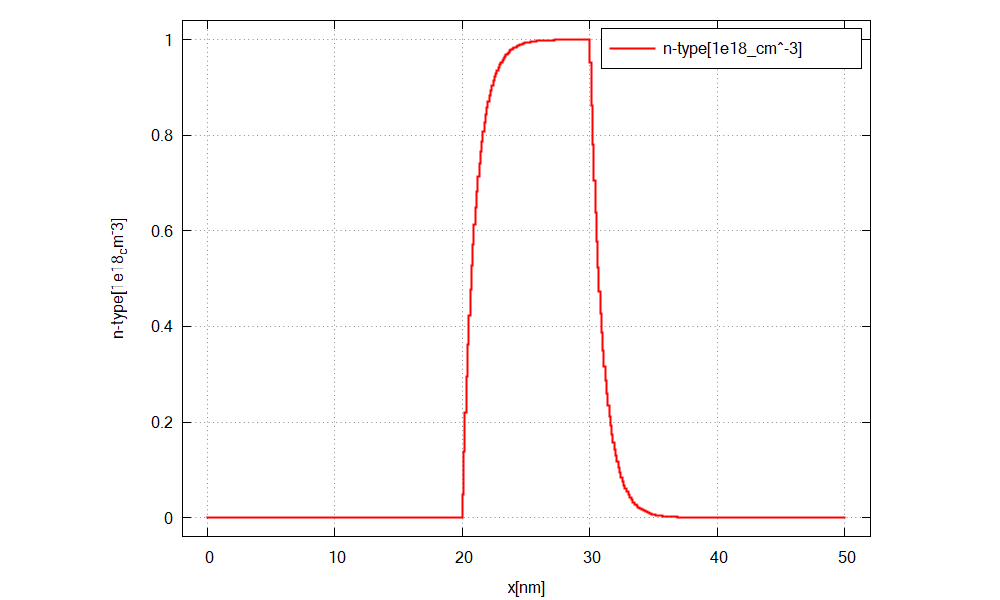
Figure 2.4.19 The doping concentration of donors along the x direction.¶
Important things to remember¶
before importing and using our own functions, we first have to define them in the
import{ }group
Last update: 16/07/2024